多光子荧光显微技术简介
多光子荧光显微技术是一件强大的科研工具,其将激光扫描显微技术的高级光学技术与长波多光子荧光激发技术相结合,可采集经高度精细的荧光团标记的试样的高分辨率三维图像。

图1.多光子激发荧光显微镜的的配置
该方法对研究活细胞和组织中动态过程的细胞生物学家特别有用,通常不会对试样造成严重的甚至破坏性的损伤。尽管经典宽场荧光显微技术通常可为活体系统中的生化事件提供亚微米级分辨率,但该技术在灵敏度和空间分辨率方面受到焦平面上下方整个区域中次生荧光引发背景噪音的限制。
多光子显微技术中的激发仅发生在衍射受限显微镜的焦点处,具有对厚生物试样进行光学切片以获得三维分辨率的能力。在x-y平面上以光栅扫描试样的方式获取单个光学切片并在连续z位置上连续扫描试样,即可合成完整的三维图像。因为可以精确地确定和控制焦点位置,多光子荧光可用于探测试样表面下的选定区域。对于连接到试样上的荧光团来说,高度局部化的激发能量可最大限度地减少光漂白及光损伤,从而提高了细胞活性,增加了后续活细胞特性研究的实验时间。此外,近红外激发波长的应用得以更深入地渗透到生物材料中,并减少较短波长下观察到的高度光散射。借助这些优势,研究人员可以在厚活体组织样品上进行实验,例如活体动物的脑切片或活体大脑以及发育中的胚胎。这些样品很难甚至不能通过其他显微技术成像。
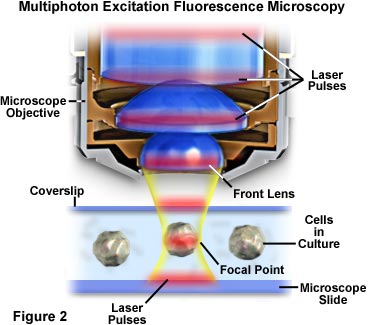
图1中的示例是多光子荧光显微技术试验中常用的配置。这款显微镜是一款直立式显微镜,可活体观察小型实验动物的活组织。显微镜机身的背面有一个钛-蓝宝石锁模脉冲激光系统,其峰值强度高,平均功率低,是多光子激发的首选激光源之一。一个经过滤的光电倍增管探测系统连接到距显微镜物镜转换器足够近的位置,可有效探测到物镜捕获的散射荧光。显微镜采集的数字图像由随附的计算机工作站进行处理和分析,从而基于光学切片完成三维重建。
传统宽场荧光显微技术受到远离焦区的次生荧光的困扰,从而会产生斑点和高背景噪音信号,常常会掩盖重要的试样细节。共聚焦显微技术利用针孔孔径从厚试样深处产生薄的(小于1微米)清晰光学切片,从而消除了焦外背景荧光,在很大程度上避免了这一问题。多光子荧光显微技术允许在更多探测中使用选择性激发,成为共聚焦显微技术的替代选择。与传统的共聚焦显微镜不同,图1所示的显微镜无需探测器附近的针孔便可获得三维分辨率,从而显著提高了发射荧光信号的效率。过去,多光子激发所需的脉冲激光系统成本高且复杂,限制了这一技术的使用。如今,交钥匙激光和商用多光子系统让多光子荧光显微技术成为许多研究的首选方法。
双光子和三光子激发
多光子激发的基本原理最早由Maria Göppert-Mayer博士在70年前撰写博士论文时提出。但是直到约30年后,脉冲红宝石激光器发明出来以后,她的假说才得到证实。在高光子密度下,两个光子可同时被吸收(由虚拟状态介导),通过组合能量来激发一个荧光团向激发态的电子跃迁。因为光子的能量与其波长成反比,所以两个光子的波长应该是单光子激发所需波长的两倍。例如,波长为640纳米(红色光)的两个光子可在组合后激发320纳米区(紫外)中吸收紫外线的荧光团,导致波长更长(蓝色或绿色)的次生荧光发射。这一独特的应用意味着可以方便地使用延伸到红外区的较长波长,在单个量子事件中激发生色团,随后在较低波长下发射次生辐射。
因为每个激发事件需要两个光子,所以要求有一个基于激发强度平方的速率常数。尽管光子不必波长相同即可诱导多光子激发,但大多数实验系统都设计有单一激光源,所以这两个光子通常是波长分布较窄的已定义光子布居的成员。与单光子吸收的情况不同,给定荧光团同时吸收两个光子的概率是入射光子间空间和时间重叠的函数。基于每个荧光团暴露于相同激光截面假设的计算表明,光子必须相继在10(-18)秒(一阿秒)内到达。此重叠期的时间尺度与中间虚拟状态的寿命(10(-17)秒或0.01飞秒)一致。
多光子荧光需要高光子密度才能确保足够水平的荧光团激发。事实上,光子浓度必须大约是同等单光子吸收数所需浓度的一百万倍。这可通过高功率锁模脉冲激光器实现,其在脉冲峰值期间生成大功率,且平均功率很低,不会损坏试样。在恒定的平均入射激光功率水平下,激光器发射的短暂但高强度的脉冲增加了给定荧光团的平均双光子吸收概率。尽量降低平均激发功率水平会减少激发期间试样中的单光子吸收量。正是单光子激发事件导致了荧光实验中的绝大部分加热和部分光损伤。
在多光子荧光实验中,典型的脉冲激光配置使用约100飞秒(10e(-13)秒)的短占空比,重复率为80-100兆赫。此方案可在试样不受到过多热量和光损伤的情况下获得令人满意的图像采集结果。每个脉冲的时间尺度,虽然通常被称为“超短”,但仍然比双光子吸收的反应时间长四到五个数量级。双光子脉冲激发的生色团中的单重态布居与常规宽场或共聚焦荧光显微技术中获得的布居相同。因此,双光子激发后的次生荧光发射与单光子实验中的观察结果没有区别。无论是被单光子还是双光子激发事件激发,罗丹明等荧光团都会发出相同宽波长范围的次生荧光。
三光子激发是一个相关的非线性光学吸收事件,可以与双光子激发类似的方式发生。不同之处在于,三个光子必须同时与荧光团相互作用,才能诱发激发单重态的跃迁。三光子激发的好处之一是成功吸收只需要比双光子吸收高十倍的光子浓度,使得该技术对某些实验来说很有吸引力。三光子激发可比双光子吸收更好地增强z轴分辨率。这是因为与三个单个光子同时相互作用的要求导致荧光团激发的截面较小。实际上,发射波长分布集中在1050纳米的红外光激光器能够激发在紫外区吸收(约350纳米,激发波长的三分之一)的荧光团。相同的激光器可以在一半波长下(525纳米)同时激发另一个荧光团,是双标记生物实验中有用的组合。
利用较短的近红外波长(低至720纳米)时,三光子荧光可将可用荧光成像范围扩展到深紫外区。在900-700纳米的波长范围内,激光将激发在240-300纳米范围内吸收的荧光团,而这在传统显微镜光学系统中几乎无法实现。用于制造荧光物镜的玻璃对低于300纳米波长的透射率非常低,但是波长较长的红外激光辐射可以轻松通过,从而生成三光子激发。
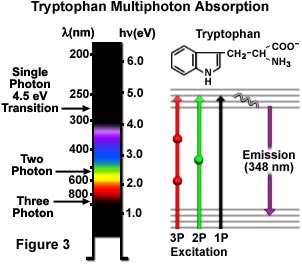
常见芳香族氨基酸色氨酸的单光子、双光子和三光子激发如图3所示。一个4.5电子伏特的单光子电子跃迁激发280纳米的色氨酸,随后在紫外区发射348纳米的次生荧光。双光子机制下的激发是通过集中在580纳米的黄绿色光完成的,而氨基酸在近红外区840纳米的辐射照射下时,会发生三光子激发。Jablonski图中(图3)显示了跃迁,其中虚拟状态由代表双光子激发的球体和代表三光子激发的两个球体表示。与其他芳香族氨基酸相比,色氨酸的荧光更强、量子产量更高,且在大多数蛋白质中仅少量存在。这些特性使得多光子显微技术成为利用色氨酸残基自发荧光的研究的一款出色工具。更高阶的非线性现象也可加以运用,包括四光子激发在内,但这些现象尚未应用于生物学研究。
双光子荧光显微技术
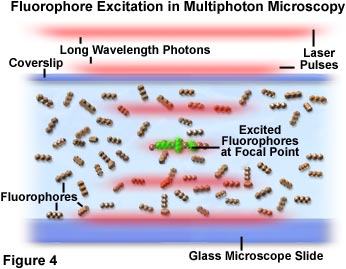
在多光子显微技术中,激发局限于焦点附近的区域,这是因为这里的光子密度最高。这一优势来自于基本的物理原理,即荧光团的双光子吸收是激发强度平方的函数。当来自脉冲激光源的光子被高数值孔径物镜聚焦时,它们会变得更加拥挤,从而增加了两个或更多个光子同时与单个荧光团相互作用的概率。显微镜焦点处光子的浓度对多光子吸收至关重要,这是唯一发生明显激发的区域。该概念如图2和图4所示,分别在宏观和微观层面上描绘了多光子激发。图2显示了显微镜物镜的放大视图,其正处于对显微镜载玻片和盖玻片间培养细胞进行成像的位置。红色激光脉冲沿物镜纵轴行进,并聚焦和集中在图中央的细胞上。
图4显示了显微镜焦点处的光子拥挤和与荧光团的相互作用。当红色激光脉冲穿过含荧光团的试样(以直线排列的三重球表示)时,激发概率在脉冲到达物镜焦点时增加。单个光子以分离成定义激光脉冲边界的漫射红线的集合体表示。图4焦区中心的一小群荧光团分子被同时吸收的两个光子激发,呈现出绿色次生荧光。焦平面外生色团吸收两个光子的概率几乎为零,这是因为该区域的光子密度不够高。
双光子激发现象之所以可能出现,不仅是因为显微镜焦点处荧光团的空间接近,还因为连续激光脉冲中包含光子的时间重叠。如上所述,双光子吸收中的激发能量与激光源产生的光子强度的平方成正比。脉冲激光束强度与距焦平面距离的平方成反比。因此,焦区附近任何位置上荧光团的激发概率与荧光团距焦平面距离的四次方成反比。脉冲激光光照锥的尺寸取决于物镜的数值孔径。因此,远离焦点的光束强度降幅与激发光锥直径的平方成正比。当光照锥扩展到焦点上下方时,荧光团的激发概率与锥体直径的四次方成反比。因此,荧光团激发仅限于焦点周围的区域,其仅代表整个试样的极薄光学切片。
激光脉冲的持续时间通常在大约100飞秒到1皮秒(10e(-13)到10e(-12)秒)的范围内,从宏观角度来看,是超短的。然而,在光子吸收事件的时间尺度上(约千分之一飞秒),脉冲的持续时间实际相当长。这就限制了荧光团饱和,并给分子足够的时间在另一轮激发前返回脉冲之间的基态。脉冲重复率范围约为80-120兆赫(MHz),为激发提供了高瞬时峰值功率,然后是平均10纳秒的驻留时间。因为典型荧光团的荧光寿命仅有几纳秒,所以激发后的分子群有足够的时间在脉冲间放松。相对短的脉冲占空比(脉冲持续时间除以脉冲间的时间)将平均输入激光功率限制为仅略大于激光扫描共聚焦显微技术的常用值。
局限于焦平面附近的双光子激发让多光子显微技术比共聚焦显微技术拥有更明显的优势。在共聚焦显微技术中,整个试样都被激发出荧光,但探测器收集的次生荧光则被共聚焦针孔限制在物镜的焦平面。这样会降低来自其他焦平面的背景噪音或荧光量,使得这些焦平面不会增加数据中的背景噪音。相比之下,多光子显微技术仅在焦平面产生荧光激发(随后生成荧光发射),这消除了背景信号,也无需共聚焦针孔。共聚焦显微技术和多光子显微技术中激发模式之间的显著差异如图5所示。图中显示了各技术的光漂白曲线。
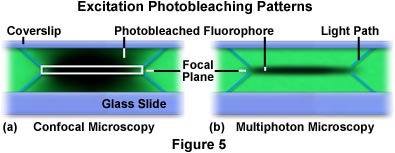
图5中显示了在用荧光团罗丹明(绿色染料)染色的方华聚合物薄膜中,重复扫描单一x-y平面后所产生的x-z光漂白图案。左侧(图5(a))显示的是由共聚焦显微镜扫描染色膜生成的曲线。扫描片中心的白色矩形代表通过针孔并由探测器成像的焦平面。矩形上下角投射的蓝色对角线代表激发光束穿过薄膜时的光路。当光束以光栅扫描方式扫描薄膜时,荧光染料被激发并发出次生荧光。最终,光漂白现象发生,用焦区中的暗区表示。在用共聚焦显微镜扫描的薄膜中(图5(a)),整个激发路径中焦平面上下方的整体激发几乎相等。与之相比,多光子显微镜产生的x-z重复扫描激发曲线将激发和光漂白限制在焦平面(图5(b))。与图5(a)中的示例类似,从焦平面发出的蓝色对角线描绘了激发光到达焦平面的路径。
多光子显微技术的局部激发具有许多优点。其中最重要的可能是,该技术可以提供较高的三维分辨率,与理想情况下共聚焦显微镜的分辨率相同。此外,焦平面外荧光团的吸收不足使得更多的激发光能够穿透试样并到达焦平面。其结果是聚焦光束穿透到试样深处的能力显著提高,通常可达共聚焦显微技术观察深度的两到三倍。
如前所述,焦区外多光子吸收的概率与沿光轴(z方向)距离的四次方成反比。当均匀分布的荧光团在高数值孔径物镜(1.4)下受到多光子激发时,约80%的吸收发生在叫做焦体积的严格受限空间内。该体积的尺寸取决于物镜的数值孔径,但对于近红外波长下的典型大孔径荧光物镜来说,该区域由一个横向直径0.3微米和轴向长度1微米的椭球体限定。
在图5(b)所示的多光子显微技术中,光漂白量(以及细胞和组织相关光损伤)的显著减少明显低于共聚焦显微技术。光漂白和光损伤是活细胞、组织和其他生物体研究中影响荧光显微技术应用的两个最重要限制因素。荧光团的激发导致基态电子向激发的单重态能量状态跃迁。在激发态的振动弛豫过程中,可能会发生到三重态的系间窜越,而不是典型的衰变回到单重基态。三重态具有极强的活性和相对较长的持续时间,使得处于这种状态的荧光团有时间与活细胞反应或发生分子降解或重排为非荧光分子。此外,处于三重态的激发荧光团可以生成单重态氧,其可与邻近生物分子上的多种官能团发生反应。在到达焦点的过程中,激发光必须在所有焦平面上穿透试样,且大部分光会在穿过焦区后继续传播相当长的距离。因此,在整个光束路径中激发的荧光团群(如在宽场和共聚焦显微技术中)将接受大量的光漂白,从而产生细胞和组织损伤,而这些损伤可通过多光子技术避免。
尽管我们对光引发细胞损伤的确切机制了解甚少,但已经证实的是减少光损伤将显著延长荧光显微技术研究中生物样品的活性。仅暴露于长波可见光和近红外光可能不会影响细胞的活性。因此,多光子显微技术相关的大部分损伤主要来自激发,且局限于焦平面。
多光子显微技术的探测器
多光子显微技术中,由次生荧光发射的光子几乎完全来自物镜焦平面,因而无需解扫探测,并可更灵活地探测几何结构。与共聚焦显微技术相比,多功能性的增加可显著提高荧光检测的效率。在采用解扫探测的系统中,由物镜收集的光在通过针孔到达探测器之前会经过一系列扫描振镜的表面反射。在提高图像分辨率的同时,共聚焦针孔会导致检测效率大幅降低,且让试样在入射光下暴露的时间更长,从而增加光损伤和光漂白的可能性。
为了最大限度地提高探测效率,非解扫探测器通常位于物镜附近,且光路的直径必须更大,才能有效地探测来自样品内部深处的散射荧光信号。多光子显微技术中常用的探测器之一是光电倍增管(PMT)。使用磷砷化镓(GaAsP)PMT探测器时,可获得比标准多碱PMT更高的量子效率,即使在微弱的荧光下也可获得高信噪比的图像。化学检测是利用电压固定细胞中的离子电流来生成受体分布的图像。

图6.
多光子显微技术中的分辨率
多光子显微技术中的分辨率不会超过共聚焦显微技术。事实上,利用较长波长(红光到近红外光;700-1200纳米)可导致多光子激发的点扩散函数更大,从而意味着横向和轴向分辨率都略有降低。例如,对于700纳米激发波长和数值孔径1.3的物镜来说,观察到的横向分辨率约为0.2微米,相应的轴向分辨率为0.6微米。当与斯托克位移大小相结合时,这些值的范围可比相同条件下常规共聚焦显微技术观察到的分辨率大30%。在实践中,有限的针孔孔径、色差和光学系统的不完美对准会降低共聚焦分辨率,这些都会缩小共聚焦和多光子显微技术间分辨率的差异。通过上述内容可以很明显的看到,当共聚焦显微技术无法对一个结构提供足够的分辨率时,多光子激发成像的效果也不会更好(甚至可能更糟)。
在以三维空间分辨率采集数字图像或计数光子时,必须区分发生在焦体积内的荧光发射和源自背景的荧光发射。对这两个信号的区分可通过仪器(共聚焦或多光子显微镜)或三维数据集的反卷积来实现。区分焦平面上荧光发射和背景荧光的能力由信背比(S/B)定义,其中S是焦平面上收集到光子的数量或强度,而B代表源自背景(焦外平面)的光子。在共聚焦扫描显微技术中,高S/B比由共聚焦针孔拒绝背景信号而产生。但在多光子激发中,S/B比必然会更大,这是因为焦平面外的激发很少。执行共聚焦计算时,可通过考虑无限小的针孔来比较多光子和共聚焦技术间的分辨率计算结果。这两种技术的信背比通常比传统宽场荧光显微技术高几个量级。
需要考虑的另一点是多光子激发允许利用在低波长紫外区中有吸收跃迁的荧光团。由于共聚焦显微技术在激发340纳米以下荧光团的能力方面有一定限制,研究人员倾向于使用波长更长、分辨率相应更低的荧光探针。在关键应用中,可通过利用共聚焦针孔以及利用空间分辨探测系统(例如放置在扫描图像平面中的CCD光电二极管阵列)限制成像波长来提高多光子显微技术的分辨率。
荧光团的激发特征
多光子研究中使用的荧光团应与单光子研究中的一样受到密切的监督。荧光探针应在方便使用的波长下具有大吸收截面、高量子产量、低光漂白速率以及尽可能低的化学和光化学毒性。荧光团还应能够承受来自激光源的高强度照射,而不会显著降解。在大多数情况下,研究人员会使用相同的常用荧光团进行双光子实验,而这些荧光团已在宽场和共聚焦荧光显微技术中广泛作为标记物使用。
常用荧光团的激发光谱是激发模式和入射光子波长的函数。由于这种依存关系,双光子吸收光谱可能(且经常)与相应的单光子光谱显著不同。在实验中,大多数已经检测的荧光团能够以其单光子最大吸收波长的两倍吸收双光子激发。尽管如此,并不能仅仅通过检查单光子截面来定量预测复杂荧光团的双光子激发光谱。高共轭非对称分子的单光子与双光子激发光谱间经常存在显著差异,从而在分子光谱中经常用于提供关于激发态结构的信息。其中一个很好的示例是芳香族氨基酸的衍生物酪氨酸和苯丙氨酸,其复杂的双光子截面与单光子激发完全不同。相比之下,色氨酸的双光子光谱(图2)与单光子激发的曲线非常相似。
在定量三维重建和反卷积实验中,应测量荧光团的双光子吸收光谱,以确保激发波长集中在吸收带中的峰值附近。虽然可以计算双光子截面,但计算过程非常复杂。尽管直接实验测量吸收光谱是首选的计量方法,但因吸收的入射功率与光源的光强波动相比较小,所以实验本身相对困难。目前,已将热透镜和声光技术用于确定吸收截面,但或许更简单的方法是检查已知量子产量的荧光团的光子发射。在设计新的双光子实验时,应检查吸收峰接近预期激发波长一半的多种荧光团。
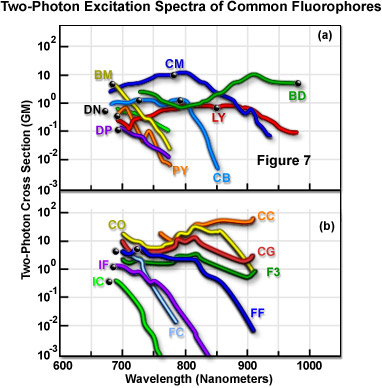
图7显示了多种常用荧光团的测得双光子荧光激发光谱的特征。图7中的数据代表双光子作用截面,由荧光发射量子效率和双光子吸收截面相乘得出。光谱使用锁模钛-蓝宝石激光器发射的线性偏振光记录。每个光谱中,黑点代表荧光团单光子最大吸收波长的两倍。表1是图7中各光谱旁两字母名称代码的图例。曲线代表荧光团双光子激发的光谱截面。
荧光团的双光子荧光激发光谱
| 荧光团名称(缩写) | 激发波长(纳米) |
|---|---|
| (BM)p-双(邻甲基苯乙烯基)苯 | 691 |
| (CB)Cascade Blue酰肼三钠盐 | 750 |
| (YL)Lucifer Yellow CH铵盐 | 860 |
| (BD-氟硼荧)4,4-二氟-1,3,5,7,8-五甲基-4-硼-3a,4-二氮杂茚-2,6-二磺酸二钠盐 | 920 |
| (DP-DAPI,非结合 DNA)4’,6-二氨基-2-苯基吲哚盐酸盐 | 700 |
| (DN-丹酰)5-二甲氨基萘-1-磺酰肼 | 700 |
| (PY)1,2-双-(1-芘癸酰基)-sn-甘油-3-磷酸胆碱 | 700 |
| (CM)香豆素307 | 776 |
| (IC)Indo-1,有Ca++ | 700 |
| (IF)Indo-1,无Ca++ | 700 |
| (FC)Fura-2,有Ca++ | 700 |
| (FF)Fura-2,无Ca++ | 720 |
| (CG)钙绿-1,有Ca++ | 725 |
| (CO)钙橙,有Ca++ | 800 |
| (CC)钙红,有Ca++ | 850 |
| (F3)Fluo-3,有Ca++ | 800 |
表1
截面测量结果表明,双光子吸收的激发峰与单光子的曲线非常相似或存在蓝移(图7)。较短的平均波长有利于将荧光团激发与锁模激光器的可用波长范围耦合。双光子吸收光谱的另一个优势是其通常比单光子吸收光谱宽很多。这通过扩大适合激发的波长范围减少了实验限制,并增强了同时激发两个双光子截面重叠但单光子光谱广泛分离的荧光团的能力。三光子截面的测量结果通常与相应的单光子光谱非常相似。
尽管单双光子激发的吸收光谱通常不同,但其他荧光特性似乎不受影响,例如寿命、发射波长和系间窜越率。这样的相似性说明可通过线性或非线性吸收达到相同的荧光激发状态,且无论激发模式如何,荧光团在被激发后的行为都将相同。这些荧光团还支持三光子激发,使得研究人员能够在大多数多光子实验中利用成熟的比率测量和光谱方法。
多光子激发中的光损伤和热损伤
所有形式的荧光显微技术都会对活细胞造成光损伤,其程度取决于激发波长、暴露时间和作为细胞探针的荧光团的化学特性。激发光引发的损伤可分为两类:热损伤和因化学反应而造成的降解。由于荧光团激发所导致的生化反应而引发的光化学副作用尚未得到很好的理解,且在不同细胞和组织类型间差异很大。另一方面,热损伤主要源自两种机制,分别由水对单光子的吸收和焦区中荧光团的双光子吸收所引起。
在研究的大多数细胞中(尤其是哺乳动物细胞),多光子荧光中使用的固有荧光团几乎不会吸收长波近红外激发辐射。然而,细胞内以及细胞和组织周围的细胞间水分可以吸收大量的红外和近红外光,生成多余的热量,从而损伤到生物试样的活性。另一方面,在共聚焦和宽场荧光显微技术中使用较短波长的可见光和紫外线照射含水生物环境时,大量的热量不会被周围的水分子所吸收。
由于水吸收单光子而产生的加热效应沿光束路径在焦平面上下方出现。在受控的平均多光子条件下,已计算出700和1000纳米下引发的温度增幅处于0.065和1.1摄氏度之间。这些计算结果与1064纳米下光镊激光激发的热测量结果相一致。激发光束保持静止时,加热效应更大,且随时间呈对数快速上升。在多光子激发实验中,由荧光团吸收引发的加热效应主要局限于焦区。随后,热量在围绕焦体积的球形对称区域内均匀释放,所以即使在高荧光团浓度下也不会产生大量热量。
结论
多光子荧光显微技术正在成为活体实验动物细胞和组织动态成像的首选方法之一。该技术在观察样品深处细胞的动态时特别有用。此外,在多光子激发中,光漂白和光损伤等副作用很小,且仅发生在焦体积周围的邻近区域。
目前,我们对细胞光毒性的理解仍浅,但其在大多荧光显微技术中都会发生。多光子显微技术中使用的较低量子能量以及对较长波长的较低固有吸收有助于减少光对活细胞和组织的有害影响,从而为细胞动态研究打开了大门。多光子显微技术研究的主要障碍是设备成本高,特别是双光子和三光子激发所需的锁模脉冲激光系统。最常用的超快激光系统是钛-蓝宝石激光器。钛-蓝宝石脉冲激光(700至1300纳米)的波长可调谐性使其可提供特别丰富的功能。这样的多功能性将促进这一技术在整个生物科学中的广泛应用。
对不起,此内容在您的国家不适用。